
Project 1 | Project 2 | Project 3 | Project 4
Project 1: Dissecting the aging-associated decline in cellular proteostasis
Project Leader: Judith Frydman (Stanford)
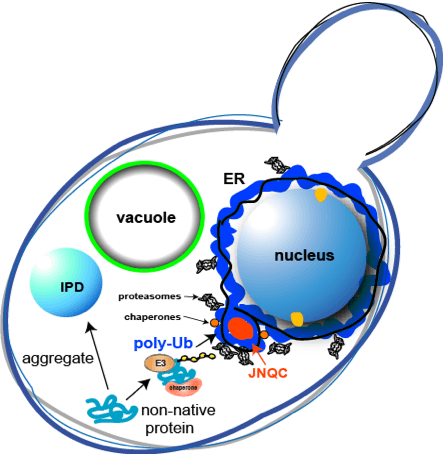
The ability to maintain a functional and healthy proteome requires cells to maintain proteostasis. Yet, this ability declines during the process of aging. Such a collapse in proteostasis results in the accumulation of misfolded and aggregated proteins that affect cell viability and are a hallmark of age-related diseases. It remains largely unknown what cellular changes are responsible for the loss of proteostasis and the accumulation of damaged proteins during aging. We propose to define the mechanisms and consequences of this proteostasis decline in order to better understand what cellular interventions could improve the aging process and ameliorate age-related diseases. Proteostasis is maintained through the interplay of molecular chaperones, which are essential for protein folding and function, and quality control factors, including the ubiquitin-proteasome system and autophagy, which target misfolded proteins for elimination. Accumulating evidence suggests that the proteostasis balance is disrupted during aging. This project will examine several phases that regulate the life cycle of a protein, including translational fidelity, chaperone function, and misfolded protein management, to determine what cellular changes dictate the widespread proteostasis collapse associated with aging. We will test two non-exclusive hypotheses for proteostasis dysfunction with age: (i) that the fidelity of cotranslational protein folding declines with age and (ii) that the capacity to manage and clear misfolded proteins declines with age. Of relevance to Alzheimer’s and other late onset diseases, we will also examine a third hypothesis: (iii) that the presence of aggregation-prone amyloidogenic protein species such as A-ß; tau and polyQ-expanded Huntingtin exacerbates these defects and accelerates the age-dependent collapse of proteostasis. We will use S. cerevisiae as the primary model for initial studies, and then examine the robustness of the aged proteostasis network in C. elegans, murine and human neuronal model systems with the Morimoto, Finley, and Finkbeiner groups, respectively. We will also investigate the effects of environmental and chemical perturbations together with the Morimoto, Finley and Kelly groups to discover fundamental mechanistic insights that can be translated into the more complex networks of C. elegans, mice and patient-derived neurons.
Specific Aims:
- Aim 1: To develop proteostasis reporters for mechanistic insight on chaperone function in aging and neurodegeneration.
- Aim 2: To develop new quantitative imaging modalities to give an aging diseased cell (yeast to human neurons) a “proteostasis physical”.
- Aim 3: To harness the cellular proteostasis network strategies to protect and repair.
Project 2: The proteasome as a modulator of neurodegenerative disease
Project Leader: Dan Finley (Harvard)
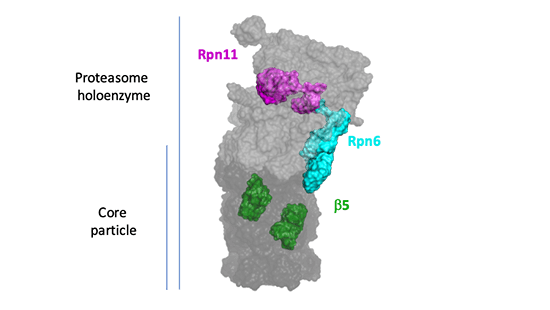 The proteasome mediates global control of protein stability and homeostasis. Historically, the proteasome has not been thought to be critical in determining the overall output of the ubiquitin-proteasome pathway (UPS), though only on the basis of supposition. We now understand that the proteasome is the focal point of general regulation of the UPS. This reflects that proteasome activity levels control protein breakdown rates and can in general be up-regulated without toxicity. Cells in tissue culture are reported to be more robust against proteotoxic challenge when proteasomes are up-regulated. These observations may be highly relevant to aging-associated disease in humans. In particular, proteasome activity declines substantially with age in the brain, motor neurons, and in other tissues and cell types. Extensive work has shown that proteasome activity is compromised in many neurodegenerative diseases. However, for a deeper and more reliable understanding of these issues, it is essential to develop in vivo mammalian model systems. Here we propose to: (i) generate transgenic mice with elevated proteasome levels, (ii) assess the resulting effects of elevated proteasome levels on health and aging of animals, and (iii) examine potential benefits of enhanced proteasome activity on Tauopathies and specific forms of ALS. Throughout these Aims, our most important tool will be global proteomics via TMT-MS3 through Core C. This method will provide precise quantitative comparisons of 8000 or more proteins in mutant and wild- type settings. Thus, the effects of the transgenic constructs on proteasome levels, protein degradation, and the chaperone network will be systemically determined at high resolution. The accumulation of disease-associated isoforms of mutant proteins will be closely followed, as well as proteome-wide signatures that may indicate a broader compromise of proteostasis in the disease model.
The proteasome mediates global control of protein stability and homeostasis. Historically, the proteasome has not been thought to be critical in determining the overall output of the ubiquitin-proteasome pathway (UPS), though only on the basis of supposition. We now understand that the proteasome is the focal point of general regulation of the UPS. This reflects that proteasome activity levels control protein breakdown rates and can in general be up-regulated without toxicity. Cells in tissue culture are reported to be more robust against proteotoxic challenge when proteasomes are up-regulated. These observations may be highly relevant to aging-associated disease in humans. In particular, proteasome activity declines substantially with age in the brain, motor neurons, and in other tissues and cell types. Extensive work has shown that proteasome activity is compromised in many neurodegenerative diseases. However, for a deeper and more reliable understanding of these issues, it is essential to develop in vivo mammalian model systems. Here we propose to: (i) generate transgenic mice with elevated proteasome levels, (ii) assess the resulting effects of elevated proteasome levels on health and aging of animals, and (iii) examine potential benefits of enhanced proteasome activity on Tauopathies and specific forms of ALS. Throughout these Aims, our most important tool will be global proteomics via TMT-MS3 through Core C. This method will provide precise quantitative comparisons of 8000 or more proteins in mutant and wild- type settings. Thus, the effects of the transgenic constructs on proteasome levels, protein degradation, and the chaperone network will be systemically determined at high resolution. The accumulation of disease-associated isoforms of mutant proteins will be closely followed, as well as proteome-wide signatures that may indicate a broader compromise of proteostasis in the disease model.
Specific Aims:
- Aim 1: To construct and characterize a panel of mouse mutants with inducible transgenes (Rpn11, Rpn6, b5) that elevate proteasome levels.
- Aim 2: To determine the effect of elevated proteasome levels on the health and aging of transgenic mice.
- Aim 3: To test for effects of elevated proteasome levels on the progression of neurodegenerative diseases.
Project 3: The autophagy lysosomal pathway:
Regulation by progranulin and its role in neurodegenerative disease
Project Co-Leaders: Steve Finkbeiner (Gladstone Institutes/UCSF), Jeff Kelly (Scripps)
Team Leader: Lisa Elia, Senior Scientist (Gladstone Institutes/UCSF)
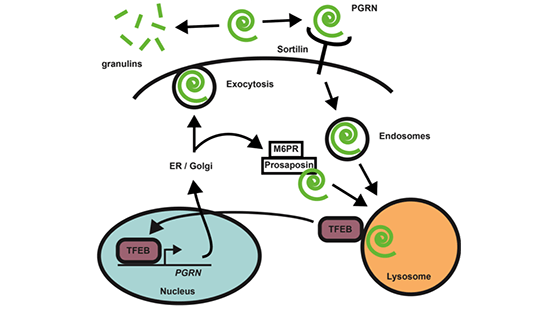 The autophagy/lysosomal pathway (ALP) is an essential component of the proteostasis network (PN). Activation of the ALP slows aging in some organisms, and mutations in genes encoding components of the pathway lead to neurodegenerative diseases, including Alzheimer’s disease, frontotemporal dementia (FTD), Parkinson’s disease, and amyotrophic lateral sclerosis (ALS). How the ALP regulates aging and neurodegenerative disease is not known. It may be mediated through the PN since the ALP is critical for degrading aggregated protein and is the only known pathway to clear damaged mitochondria and lipids. We aim to better understand mechanisms of how the ALP may affect age-dependent neurodegeneration, and propose to determine how progranulin (GRN), a polypeptide with links to both aging and increased risk of AD patients to develop the lysosomal storage disorder neuronal ceroid lipofuscinosis. Aggregation and accumulation of TDP43 into intracellular inclusions are pathological hallmarks in the CNS of 90% of ALS patients, some AD and PD cases, and 50% of FTD patients, including all due to GRN haploinsufficiency. Only rarely are mutations in TDP43 found, indicating that, in most cases, inclusions of wild-type (WT) TDP43 must form, which we believe is due to a fundamental mismatch between the production and clearance of misfolded TPD43. We hypothesize that GRN deficiency leads to neurodegeneration by causing dysfunction of ALP and toxic accumulation of TDP43. Here, we propose to: (i) determine how GRN deficiency affects the macromolecular composition of lysosomes and on the PN with RNAseq, proteomics and lipidomics in vivo and in human neurons from FTD patient i-neurons, and to quantify the dynamic response of the PN in GRN-deficient FTD i-neurons to stress with longitudinal single measurements of the ALP, the UPS and the unfolded protein response, (ii) define the role of GRN in the proteostasis of neurodegenerative disease-causing proteins by measuring the effect of GRN deficiency and rescue on the nuclear localization, phosphorylation, half-life, aggregation and neurotoxicity of mutant TDP43, and by surveying the effects of GRN deficiency and rescue on the turnover of tau, synuclein and mHTT, and (iii) discover, validate and characterize new chemical probes to modulate autophagy, mitophagy, and other dimensions of the ALP.
The autophagy/lysosomal pathway (ALP) is an essential component of the proteostasis network (PN). Activation of the ALP slows aging in some organisms, and mutations in genes encoding components of the pathway lead to neurodegenerative diseases, including Alzheimer’s disease, frontotemporal dementia (FTD), Parkinson’s disease, and amyotrophic lateral sclerosis (ALS). How the ALP regulates aging and neurodegenerative disease is not known. It may be mediated through the PN since the ALP is critical for degrading aggregated protein and is the only known pathway to clear damaged mitochondria and lipids. We aim to better understand mechanisms of how the ALP may affect age-dependent neurodegeneration, and propose to determine how progranulin (GRN), a polypeptide with links to both aging and increased risk of AD patients to develop the lysosomal storage disorder neuronal ceroid lipofuscinosis. Aggregation and accumulation of TDP43 into intracellular inclusions are pathological hallmarks in the CNS of 90% of ALS patients, some AD and PD cases, and 50% of FTD patients, including all due to GRN haploinsufficiency. Only rarely are mutations in TDP43 found, indicating that, in most cases, inclusions of wild-type (WT) TDP43 must form, which we believe is due to a fundamental mismatch between the production and clearance of misfolded TPD43. We hypothesize that GRN deficiency leads to neurodegeneration by causing dysfunction of ALP and toxic accumulation of TDP43. Here, we propose to: (i) determine how GRN deficiency affects the macromolecular composition of lysosomes and on the PN with RNAseq, proteomics and lipidomics in vivo and in human neurons from FTD patient i-neurons, and to quantify the dynamic response of the PN in GRN-deficient FTD i-neurons to stress with longitudinal single measurements of the ALP, the UPS and the unfolded protein response, (ii) define the role of GRN in the proteostasis of neurodegenerative disease-causing proteins by measuring the effect of GRN deficiency and rescue on the nuclear localization, phosphorylation, half-life, aggregation and neurotoxicity of mutant TDP43, and by surveying the effects of GRN deficiency and rescue on the turnover of tau, synuclein and mHTT, and (iii) discover, validate and characterize new chemical probes to modulate autophagy, mitophagy, and other dimensions of the ALP.
Specific Aims:
- Aim 1: To investigate mechanisms of frontotemporal dementia.
- Aim 2: To discover and develop new small molecule autophagy inducers as possible treatments.
Project 4: Organismal proteostasis in aging and neurodegenerative diseases
Project Leader: Richard Morimoto (Northwestern)
Team Members: Anan Yu, Research Assistant Professor (Northwestern), Laura Bott, Research Assistant Professor (Northwestern), Sue Fox, Research Lab Manager (Northwestern), Alejandro Rodriguez Gama, Postdoctoral Fellow (Northwestern)
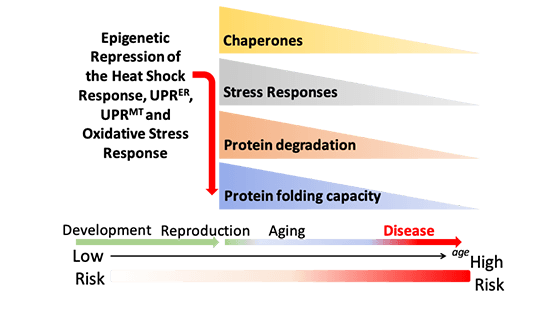 Aging affects intertissue stress signaling and functional properties of the proteostasis network (PN) influencing the regulation of organismal proteostasis and therefore the course of misfolding and aggregation of Tau, SOD1 and polyglutamine proteins associated respectively with Alzheimer’s disease, ALS, and Huntington’s disease. Each tissue in a metazoan contains a unique proteome whose integrity relies on the PN consisting of molecular chaperones, transport, and clearance pathways that determines cellular function and organismal health. Our group is interested in the characteristics and regulation of the PN for optimal quality control in a complex metazoan, and in the mechanisms that go awry resulting in PN failure during stress, aging, and neurodegenerative diseases. We have previously demonstrated that the heat shock response (HSR) in C. elegans is under cell-nonautonomous control by specific sensory neurons that regulate induction of the HSR in somatic tissues. We have also shown that tissues communicate the functional status of their internal folding environment to one another via transcellular chaperone signaling. The composition, activity, and dynamics of the PN throughout the lifespan of an organism remain unknown. We will characterize the components and the functionality of the PN in different tissues during development and aging, determine how stress resilience of individual cell types is affected by aging, and examine how aging affects cell-nonautonomous stress signaling and proteostasis in C. elegans. We will characterize different arms of the PN during aging and in animals expressing Tau, SOD1, and polyglutamine proteins using proteostasis reporters (Core B) and transcriptome profiling in collaboration with Projects 1-3, genetically re-engineer the proteasome to reverse age-dependent changes in its activity (Project 2), and test small-molecule approaches to modulate the PN with Core D, with the overall goal to suppress aggregation, restore organismal proteostasis, health, and longevity.
Aging affects intertissue stress signaling and functional properties of the proteostasis network (PN) influencing the regulation of organismal proteostasis and therefore the course of misfolding and aggregation of Tau, SOD1 and polyglutamine proteins associated respectively with Alzheimer’s disease, ALS, and Huntington’s disease. Each tissue in a metazoan contains a unique proteome whose integrity relies on the PN consisting of molecular chaperones, transport, and clearance pathways that determines cellular function and organismal health. Our group is interested in the characteristics and regulation of the PN for optimal quality control in a complex metazoan, and in the mechanisms that go awry resulting in PN failure during stress, aging, and neurodegenerative diseases. We have previously demonstrated that the heat shock response (HSR) in C. elegans is under cell-nonautonomous control by specific sensory neurons that regulate induction of the HSR in somatic tissues. We have also shown that tissues communicate the functional status of their internal folding environment to one another via transcellular chaperone signaling. The composition, activity, and dynamics of the PN throughout the lifespan of an organism remain unknown. We will characterize the components and the functionality of the PN in different tissues during development and aging, determine how stress resilience of individual cell types is affected by aging, and examine how aging affects cell-nonautonomous stress signaling and proteostasis in C. elegans. We will characterize different arms of the PN during aging and in animals expressing Tau, SOD1, and polyglutamine proteins using proteostasis reporters (Core B) and transcriptome profiling in collaboration with Projects 1-3, genetically re-engineer the proteasome to reverse age-dependent changes in its activity (Project 2), and test small-molecule approaches to modulate the PN with Core D, with the overall goal to suppress aggregation, restore organismal proteostasis, health, and longevity.
Specific Aims:
- Aim 1: To examine the effects of aging and proteotoxic stress on PN composition and function in C. elegans during aging and stress. We will employ reporters to relate PN functionality to PN composition in tissues in short-and long-lived animals.
- Aim 2: To examine how Tau, SOD1, and polyglutamine proteins affects intertissue stress signaling in neurons, muscle, intestine to identify tissue circuits and directionality of stress signaling across tissues during aging.
- Aim 3: To use proteostasis regulators to reset and restore the organismal PN, stress resilience and to counteract the age-dependent decline in proteostasis and prevent proteotoxicity.
